

Published: 4th Aug, 2022
Context
Deep Mind, a company owned by Google, announced that it had predicted the three-dimensional structures of more than 200 million proteins using Alpha Fold.
Background
- AlphaFold 1(2018) was built on work developed by various teams in the 2010s, that looked at the large databanks of related DNA sequences now available from many different organisms (most without known 3D structures), to try to find changes at different residues that appeared to be correlated, even though the residues were not consecutive in the main chain.
- Such correlations suggest that the residues may be close to each other physically, even though not close in the sequence, allowing a contact map to be estimated.
- It also used more advanced learning methods than previously to develop the inference.
- Combining a statistical potential based on this probability distribution with the calculated local free-energy of the configuration, the team was then able to use gradient descent to a solution that best fitted both.
Analysis
What is AlphaFold?
- AlphaFold is an AI-based protein structure prediction tool.
- It is based on a computer system called a deep neural network.
- Inspired by the human brain, neural networks use a large amount of input data and provide the desired output exactly like how a human brain would.
- The real work is done by the black box between the input and the output layers, called the hidden networks. AlphaFold is fed with protein sequences as input.
- When protein sequences enter through one end, the predicted three-dimensional structures come out through the other.
How does the AlphaFold work?
- It uses processes based on training, learning, retraining, and relearning.
- The first step uses the available structures of 1, 70,000 proteins in the Protein Data Bank (PDB) to train the computer model.
- Then, it uses the results of that training to learn the structural predictions of proteins not in the PDB.
- Once that is done, it uses the high-accuracy predictions from the first step to retrain and relearn to gain higher accuracy of the earlier predictions.
- By using this method, alphaFold has now predicted the structures of the entire 214 million unique protein sequences deposited in the Universal Protein Resource (UniProt) database.
|
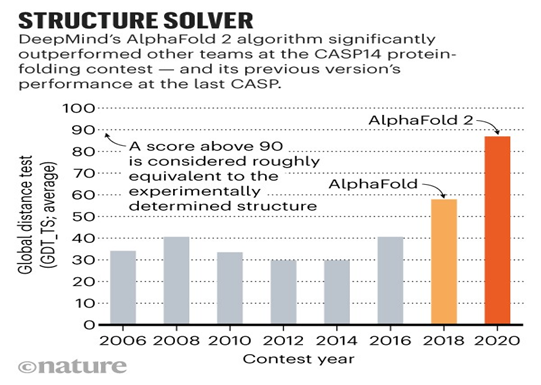
Global Distance Test
|
What is CASP14?
- DeepMind's AlphaFold placed first in the overall rankings of the 13th Critical Assessment of Techniques for Protein Structure Prediction (CASP).
- In November 2020, DeepMind's new version, AlphaFold 2, won CASP14.
- It gives ranking to the Alphafold models for predicting the structures of proteins.
What are their contributions to the Health sector?
- SARS-CoV-2: AlphaFold has been used to predict structures of proteins of SARS-CoV-2, the causative agent of COVID-19. The structures of these proteins were pending experimental detection in early 2020.
- Results were examined by the scientists at the Francis Crick Institute in the United Kingdom before being released into the larger research community.
- The team also confirmed accurate prediction against the experimentally determined SARS-CoV-2 spike protein that was shared in the Protein Data Bank, an international open-access database, before releasing the computationally determined structures of the under-studied protein molecules.
- The team acknowledged that although these protein structures might not be the subject of ongoing therapeutical research efforts, they will add to the community's understanding of the SARS-CoV-2 virus.
- Specifically, AlphaFold 2's prediction of the structure of the ORF3aprotein was very similar to the structure determined by researchers at the University of California, Berkeley using cryo-electron microscopy.
- This specific protein is believed to assist the virus in breaking out of the host cell once it replicates.
- This protein is also believed to play a role in triggering the inflammatory response to the infection.
What do these developments mean to India?
- Historical advancements: The Indian community of structural biology is strong and skilled. It needs to quickly take advantage of the AlphaFold database and learn how to use the structures to design better vaccines and drugs.
- Help in COVID and virus mutation understanding: This is especially important in the present context. Understanding the accurate structures of COVID-19 virus proteins in days rather than years will accelerate vaccine and drug development against the virus.
- Encourage the PPP model: India will also need to speed up its implementation of public-private partnerships in the sciences.
- Participation of academic institutions: Learning from this, India could facilitate joint collaborations with the prevalent hardware muscle and data science talent in the private sector and specialists in academic institutions to pave the way for data science innovations.
- Helps to develop drugs and vaccines: The Indian community of structural biology needs to take advantage of the AlphaFold database and learn how to use the structures to design better vaccines and drugs.
What are some limitations of the Alpha-fold?
AlphaFold DB currently focuses on the use case validated in CASP14: predicting the structure of a single protein chain with a naturally occurring sequence. Though has some limitations;
- The version of AlphaFold used to construct in this database does not output multi-chain predictions/ (complexes).
- In some cases, the single-chain prediction may correspond to the structure adopted in the complex.
- In other cases (especially where the chain is structured only on binding to partner molecules) the missing context from surrounding molecules may lead to an uninformative prediction.
- For regions that are intrinsically disordered or unstructured in isolation, AlphaFold is expected to produce a low-confidence prediction (pLDDT < 50), and the predicted structure will have a ribbon-like appearance.
- AlphaFold has not been validated for predicting the effect of mutations. In particular, AlphaFold is not expected to produce an unfolded protein structure given a sequence containing a destabilizing point mutation.
- Where a protein is known to have multiple conformations, AlphaFold usually only produces one of them. The output conformation cannot be reliably controlled.
- AlphaFold does not predict the positions of any non-protein components found in experimental structures (such as cofactors, metals, ligands, ions, DNA/RNA, or post-translational modifications).
Conclusion
AlphaFold 1 proved that neural networks possess the complexity required in order to be capable of modeling the protein folding mechanism. AlphaFold 2 further improves accuracy by using a more representative internal representation and embedding equivariance knowledge in the model. However, the technologies in the field of Health sector-based lab research need to focus on seeing the current scenario.